






Sindrome di Lennox-Gastaut
Sintesi della condotta assistenziale in emergenza malattie rare
Encefalopatia epilettica grave dell’infanzia, caratterizzata da crisi epilettiche multiple (assenze atipiche, convulsioni assiali toniche e cadute atoniche o miocloniche improvvise); onde lente diffuse intercriptiche all'elettroencefalogramma durante la veglia <3 Hz, picchi ritmici rapidi (10 Hz) durante il sonno; compromissione cognitiva.
L'età di esordio è tra i 2 e i 10 anni, con un picco tra i 3 e i 5 anni. Nel 50% dei casi si manifesta successivamente alla sindrome di West.
Specifiche condotte assistenziali in relazione alla patologia
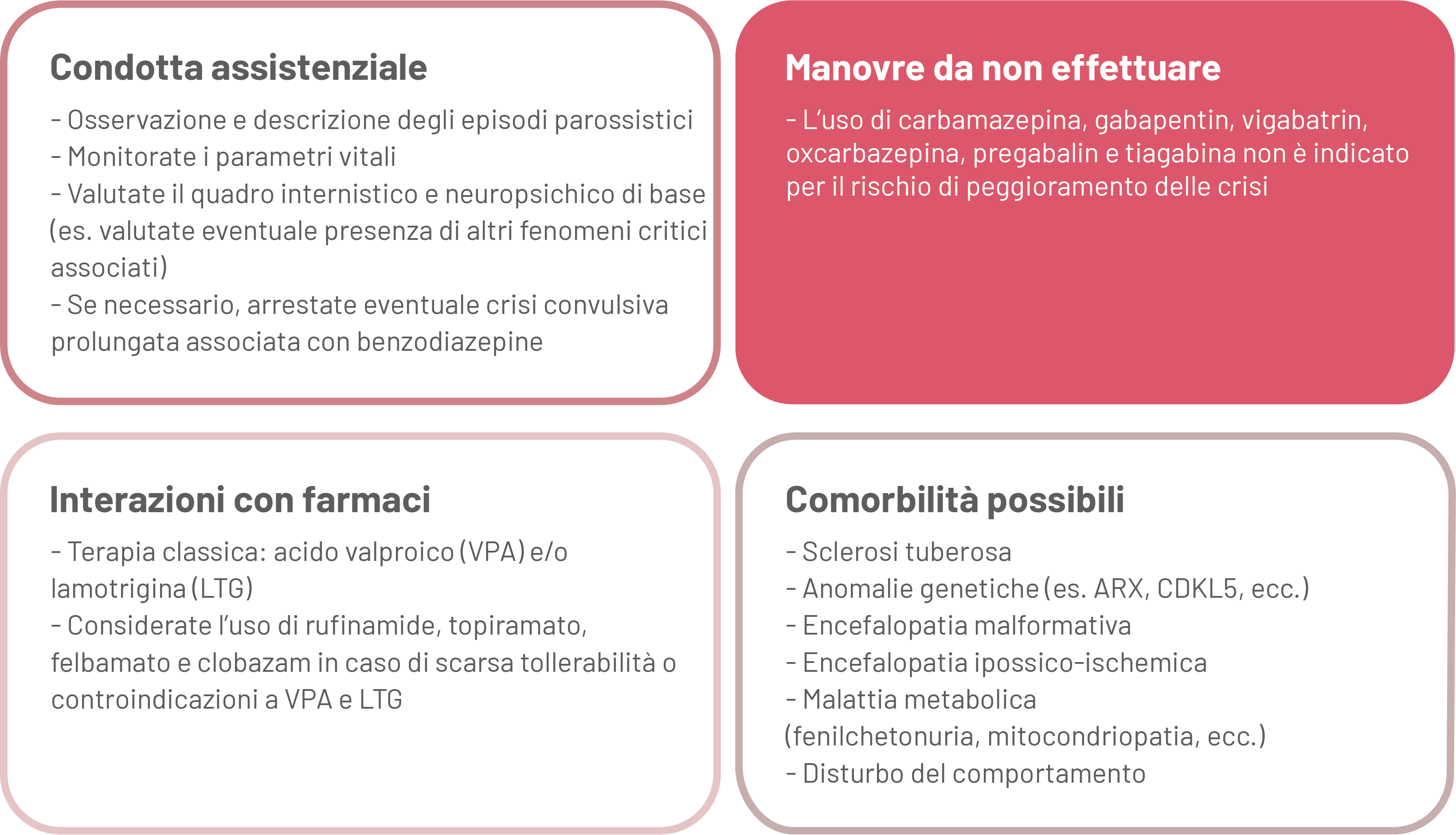
La diagnosi si basa essenzialmente sulla valutazione clinica (osservazione e descrizione degli episodi; monitoraggio dei parametri vitali e del quadro internistico e neuropsichico di base) combinata con quella elettroencefalografica. Il trattamento degli episodi critici è farmacologico, più raramente chirurgico. In emergenza, potrebbe essere necessario arrestare eventuale crisi epilettica convulsiva prolungata associata.
Manovre da non effettuare
L’uso di carbamazepina, gabapentin, vigabatrin, oxcarbazepina, pregabalin, e tiagabina non è indicato per il rischio di peggioramento delle crisi.
Interazioni con i farmaci
I due trattamenti più efficaci sono acido valproico e/o lamotrigina; va considerato inoltre l’uso di rufinamide, topiramato, felbamato e clobazam in caso di scarsa tollerabilità o controindicazioni a valproico e/o lamotrigina.
Comorbilità possibili
In un'elevata percentuale di casi sono state rilevate anomalie cerebrali. Le cause più comuni sono le malformazioni (molto spesso la sclerosi tuberosa) o i postumi di un'ischemia o una meningoencefalite, le anomalie genetiche (Sindrome di Down, delezione 1p36, mutazioni dei geni ARX, CDKL5 e STK9) o una malattia metabolica (una patologia mitocondriale o la fenilchetonuria). Frequente la comorbidità con disturbo del comportamento.